2. Tutorial (Basic Usage)¶
The MeRP pipeline works in the python console. First, from the command line, install the package.
>>> sudo python setup.py install
Then enter your python console
>>> python
To import and initialize MeRP after entering the Python console, run the commands:
>>> import merp
>>> m = merp.Merp()
In this tutorial we will address the following question:
Does LDL cholestrol levels have a causal effect on the risk of Coronary Heart Disease?
2.1. Pulling Genetic Instruments from GWAS Catalog Data¶
Run this python command first in the package directory to pull latest NHGRI GWAS catalog and create a set of IV trait files in the directory /traitFiles along with an index file mapping traits to IDs in merp directory.
>>> m.pull()
2.1.1. Optional argument:¶
Default settings presented.
- keyword = “”
- If keyword specified, only IVFs with trait/disease name containing keyword will be generated in /traitFiles.
Note
Frozen version of catalog (in /data) and trait IVFs (/traitFiles) from 8/10/14 included in package. Running pull function will overwrite these files.
2.2. Selecting/Updating IVs for Traits of Interest¶
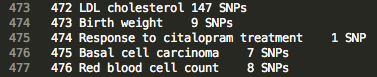
Browse the “index” file in the home directory to find ID numbers associated with traits of interest.
Example Index
Warning
MeRP pipeline is specifically designed for traits and biomarkers that are measured in natural log of odds ratio (beta) and not in odds ratio in order to run the analysis with disease endpoints. Please keep this in my mind when selecting trait of interests, and convert odds ratios to betas before analysis.
Run update function with trait file(s) of interest to add in non-risk allele and assign directional values to the beta effects according to the units description.
To update LDL cholesterol file, we run:
>>> m.update('traitFiles/472')
- Output:
- Updated file added to /traitFiles as 472_update.
- List of SNPs without allele info from 1000 genomes if applicable. Non-risk allele no reported. [NR]
Warning
While we use 1000 genomes SNP data to find non-risk alleles for SNPs, there are inevitably SNPs that are missing data. Due to this limitation, SNP lines with [NR] for non-risk allele should either be filled in with independent look up (dbSNP, etc.)or deleted before proceeding with filter().
2.3. Filter Trait IVFs for Confounding and LD¶
>>> m.filter('traitFile 472_update’,nhgri_ignore='confounders/nhgri_ignore.txt',confounders='confounders/confounders.txt',primary_confounders='confounders/prim_confounders_chd.txt']
- Output:
- Trait file filtered for confounding and correlation generated in : /traitFiles/461_updatefiltered
Note
The filter will not work if there are still [NR] non-risk alleles or other format issues. Users can input their own trait files in the filter as long as it is properly formatted. See Detailed Workflow for details on file formats.
2.3.1. nhgri_ignore and confounders¶
These two required text files should be kept in the merp directory, /confounders, and consist of a list of items separated by new lines.
nhgri_ignore.txt: list of traits that are found in NHGRI catalog that may be related to or similar to trait of interest but is not confounding.
Note
You may run filter function once with blank nhgri_similar.txt file. The program will print out exclusions because of associations in NHGRI catalog. Select those that are similar to trait of interest and that you DON’T want SNPs to be tossed out for.
confounders.txt: list of column headers from pval_file that are potentially related to trait of interest or endpoint disease. Default use is all metabolic pval file (see detailed workflow)
primary_confounders.txt (optional): list of column headers from pval_file that we would like to be more strict on. May be known to directly affect disease endpoint
Note
Package includes list of all confounders in /confounders/confounders.txt of default pval file. Optional primary confounder file is formatted the same way.
2.3.2. Optional arguments:¶
Default settings presented.
- primary_confounders = “”
- String with path/to/file
- If keyword specified, only IVFs with trait/disease name containing keyword will be generated in /traitFiles.
- verbose = True
- If False, does not print detailed logs. Text logs always generated in /logs.
- out = False
- If True, means user is bringing in outside IVFs not generated from NHGRI catalog.
- localp = False
- Default setting streams metabolic pval_file from our server. To use local copy or customized pval_file at /data/pval_file.txt, set to True.
- pmaxprimary = 0.01
- if a SNP has a single violation of p<pmaxprimary, SNP excluded
- pmax1 = 0.05
- if a SNP has more than threshold1 number of associations with p<pmax1, SNP excluded.
- threshold1 = 3
- if a SNP has more than threshold1 number of associations with p<pmax1, SNP excluded.
- pmax2 = 0.001
- if a SNP has more than threshold2 associations of pmax2, SNP excluded.
- threshold2 = 0
- if a SNP has more than threshold2 associations of pmax2, SNP excluded.
- rsq_threshold = 0.05
- SNPs with R^2 value > rsq_threshold are clustered together, with only lead SNP passing on.
- max_fraction = 0.1
- If number of total pmax1 violations exceeds max_fraction * total number of tests, then cut SNPS until below max_fraction of tests.
As shown above, LDL Cholesterol, our nhgri_ignore.txt file contains traits found in NHGRI catalog related to LDL. Our confoudners.txt file contains headers that are potential confoudners and primary_confounders.txt contains headers that are known to associate with coronary heart disease.
2.3.3. Units¶
Due to the current nature of the NHGRI GWAS catalog, there may be several different units, or variations on the same unit present in the filtered IVF. At the end of the filter, we output the different units that must be addressed before moving on to IV calculations. For accurate estimations using calc() the units must all agree. Users may acheive this by tossing out SNPs or applying appropriate conversions and manually editing beta and unit values in a text editor.
Note
The unit “units” most often means standard deviation units. As so s.d, sd, sd_%, etc. Be aware of these and use your best judgement when fixing units.
For the purposes of our tutorial, we delete SNP lines that are not in s.d units and those without non-risk allele in 1000 genomes data before proceeding to calculation.
2.4. Calculate Trait-Disease Casual Effect Estimate¶
Lastly, we calculate the estimated causal effect of LDL cholesterol on Coronary Heart Disease (CHD) using our filtered trait IVF along with the provided CVD gwas summary file.
>>> m.calc('traitFile/472_update', 'data/demo_chd.txt')
- Output:
- Summary and individual SNP result file with estimated causal effect (alpha), standard error on effect, and p-value on result.
The summary and individual result files can be found at /analysis
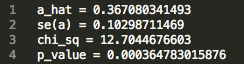
472_updatefiltered_MR_result:
472_updatefiltered_MR_result_indiv:
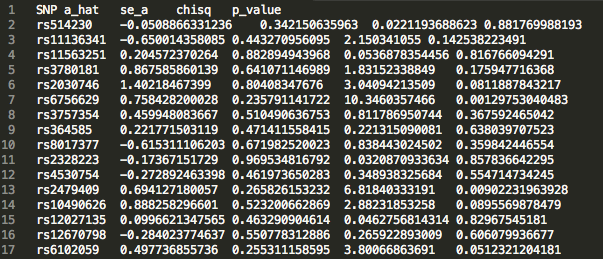
We see that there is an estimated effect of 0.367 increase in log odds ratio (a_hat) with an increase in 1 standard deviation of LDL cholesterol levels. The p value is on the order of 10 ^ -4 , indicating a significant effect. This result supports the hypothesis that LDL cholesterol levels causally increase risk for Coronary Heart Disease, matching epidemiological observations.
2.5. Optional: Export to gtx R Package for Additional Analysis¶
Users may convert the filtered files to appropriate format for gtx R package by Toby Johnson from which our statistical analysis is derived. Users may additionally run heterogeneity tests, repeat analysis, and plot results of regression using gtx_helper.R found in /src/, modifying name to match path to converted file (automatically generated in new /for_gtx directory)
>>> m.convert_to_gtx('path/to/472_updatefiltered’,'path/to/endpoint_file',dis_name = 'T2D')